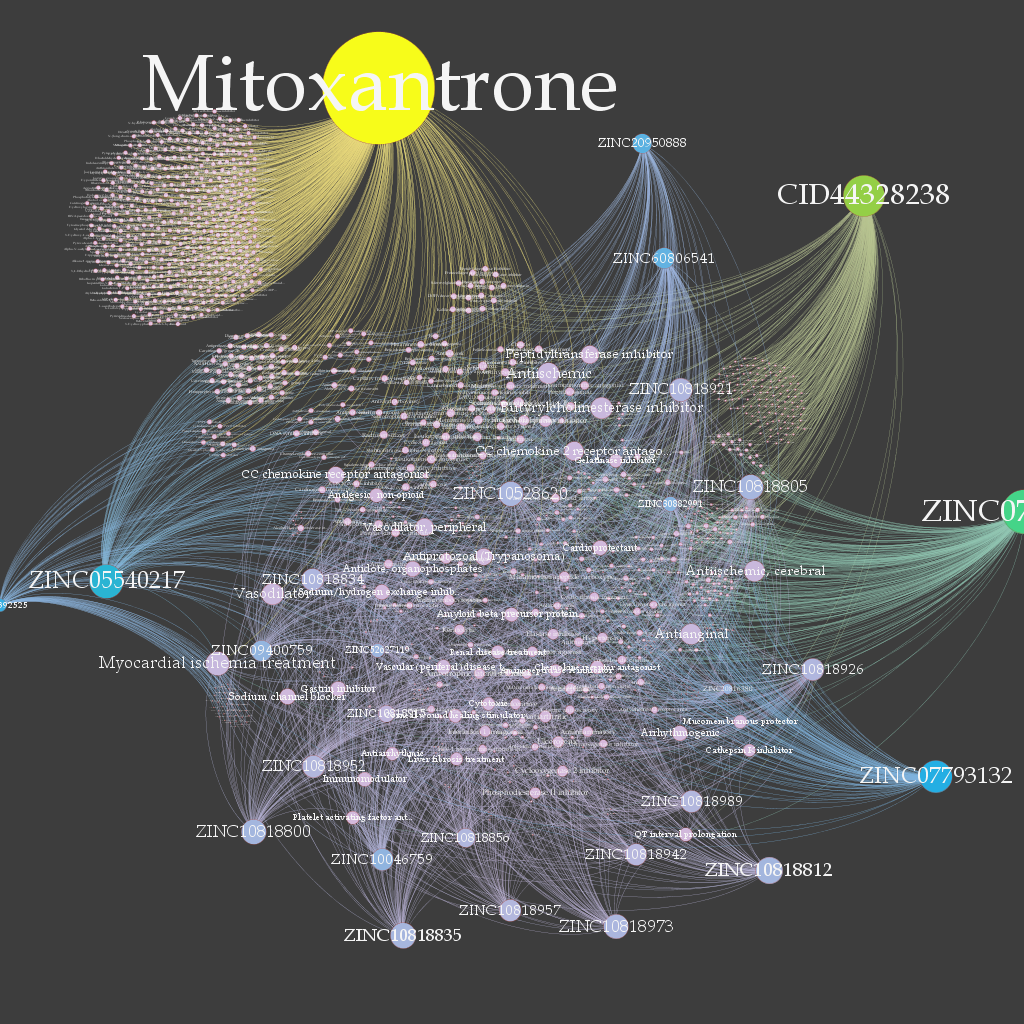
To start with i just used various screening strategies like the pharmacophore, vROCS and Ftrees algorithm to screen the kinase datasets obtained from Otava Chemical library, Kindock, our own internal dataset(chem2Bio2Rdf) and ZINC pharmer hits .I Used Omega2 to generate the conformers compounds and then use the screening softwares to screen the datasets. Then i ranked the compound based on the harmonic mean of the ranked compounds of the screen sets. .After then using pipeline pilot's different admet filters listed some compounds.
There are hits which were highly ranked in pharmacophore but didnt show any ranks in other methods.
Other methods also had some compounds which were not ranked on other methods they are given below.
Also compounds where predicted for the biological activity using the
pass prediction.Figure below shows the pass map of the biological activity with the targets.